
You’re in the final stretch of your go-to-market plan and you’re ready to label your finished medical device. Labeling is the easy part, right?
When it comes to medical devices, the term labeling encompasses much more than a sticker – it refers to a set of materials and information about your device that is crucial for patient safety and effective device use. Major regulatory bodies enforce specific requirements to ensure that medical device labeling meets certain standards. Compliance with these requirements is a legal obligation for medical device and IVD manufacturers as long as your device is on the market.
In this article, we’ll give you a high-level understanding of what labeling is, explain terminology you should know, and discuss key FDA, EU, and international requirements.
First of all, let’s establish what labeling is and what it isn’t. Labeling is written, printed, or graphic matter affixed to or associated with a medical device that is related to its identification, technical description, or use. Below is a more detailed list of what is in the scope of labeling:
Any of the product’s labeling materials can be scrutinized by regulators to ensure they comply with labeling requirements and guidance, whether they are printed or digital (i.e., e-labeling). Let’s look at how two major regulatory frameworks – the US FDA and EU Medical Devices Regulation (EU MDR 2017/745) – regulate device labels and labeling.
Title 21 of the Code of Federal Regulations is very prescriptive regarding medical device labeling. 21 CFR Part 801 covers every aspect of labeling, from unique device identification (UDI) to translations. 21 CFR Part 820 – Quality System Regulation, Section 820.120 outlines labeling controls and monitoring procedures as part of your quality management system (QMS).
In the EU MDR, labeling requirements are addressed in Annex 1, Chapter III – Requirements Regarding the Information Supplied with the Device, Section 23.
When you think about medical device labeling, the actual label on the device is probably the first thing that comes to mind. While the label is only one piece of the labeling puzzle, it is a critical piece to get right.
The term label refers to the written, printed, or graphic information appearing on the device itself, on the package of each unit, or on the packaging of multiple devices. The label can also include a marking, which is engraved or printed directly onto the product. As you’ll see in the table below, US and EU label requirements are relatively consistent.
While many fundamental label requirements are the same, it’s important to review US and EU requirements carefully to ensure that you comply, particularly if your product carries device-specific label requirements. Some discrepancies also exist between US and EU requirements for when to apply specific label exceptions.
UDI is “a series of numeric or alphanumeric characters created through a globally accepted device identification and coding standard” that allows the identification and traceability of a specific device [EU MDR Annex VI, Part C(1)]. Your device UDI must be created by an accredited issuing entity and registered in a country-specific database, such as GUDID in the US or EUDAMED in the EU.
UDI includes two parts:
UDI also includes a UDI carrier, which comprises automatic identification and data capture (AIDC) technology, such as a barcode, and, if applicable, a human-readable interpretation (HRI). The UDI carrier should be placed on the device label or on the device itself, should be included on all levels of packaging, and must be readable during normal use and throughout the device’s intended lifetime.
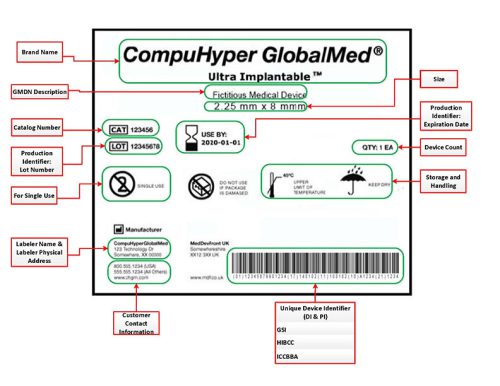
Symbols aren’t required, but they can save valuable real estate on your device label, bypass language barriers, and reduce translation needs. FDA addresses the use of symbols in 21 CFR Part 801.15. In the EU, ISO 15223-1 is the commonly used standard for compliance with the MDR. You can design your own symbols, but you will need to explain your custom symbols in accompanying text or a symbols glossary in the IFU, which is why most companies use internationally recognized symbols.
As the name implies, your device’s instructions for use explain everything the user needs to know about your device, including how and when to operate it. IFU can refer to a single, comprehensive document or several documents, including manuals, quick-start guides, online tutorials, and more. Your IFU should always include the following:
It may be tempting to conserve space (and time) when preparing your IFU, especially if you’re required to translate your IFU into multiple languages. Always prioritize clarity and attention to detail over brevity to ensure that your IFUs are accurate, thorough, and easy to understand. Well-written and organized IFUs are essential to preventing use errors and mitigating risk to patients or users.
Requirements aside, it’s also important that you consider how your IFU will be used. Here are some tips:
Translation requirements are a major point of difference between the US and EU. US FDA requires English-language labeling, with some exceptions. However, the EU MDR requires labeling translations to accommodate the official languages of the EU’s 27 Member States, which could include 24 distinct languages. In general, all labeling must be translated, including:
You will need controls in place to ensure that even minor labeling revisions are accurately reflected across all languages. Account for translation time when making any changes to labeling for your products distributed in the EU.
As far as regulators are concerned, any direct or implied assertion about your product, whether it’s in an ad or a social media post – and whether it was made by someone at your company or not – can be considered a claim. Claims that “misbrand” (FDA’s term) or misrepresent your product can violate regulator guidelines and get your device taken off the market. Therefore, any claim about your device should be carefully crafted and monitored.
Complying with device labeling requirements calls for meticulous attention to detail. If you’re not sure where to start, our in-depth class on FDA and EU Medical Device Labeling Requirements will provide clarity on the path forward. You’ll learn how to tackle all aspects of labeling for your product, including requirements for specific device types, usability and risk considerations, and how to proceed with your FDA 510(k) or EU submission.